
Storage of HSV in Saline Leads to Membrane Disruption and Endothelial Dysfunction
Christy Guth, Kalyan Chadalavada, Padmini Komalavilas, Joyce Cheung-Flynn, Colleen Brophy
Vanderbilt University Medical Center, Nashville, TN
Background: Human saphenous vein (HSV) is the most common conduit for vascular bypass grafting. HSV is often stored in heparinized saline solution (HS) after harvest and prior to implantation. Storage in HS impairs HSV endothelial function and increases vein graft failure. In spite of this, many surgeons continue to store HSV in HS after harvest and prior to implantation.
Heparin is a highly negatively charged polysulfated molecule and normal saline does not contain a buffer. The pH of HS varies between 5.0 and 6.0. Lowering the pH of buffered solution led to impaired endothelial function of HSV suggesting that acidosis is the cause of injury with NS. However, the mechanism by which acidosis leads to impaired endothelial function is not known.
To increase understanding of vein graft failure due to endothelial injury, this study sought to determine the mechanism by which storage in HS impairs endothelial function.
Methods: Primary HSV endothelial cells (HSVEC, PromoCell, Heidelberg, Germany) were grown in Endothelial Cell Growth Medium (PromoCell), maintained in a 37C and 5% CO2 incubator and were passaged at 80% confluence. Cells between passages 2 and 4 were used in the experiments. The cells were incubated in culture medium (control), heparinized (10U/ml) saline (HS), or heparinized PlasmaLyte (HP) for 2 hours (pH 7.0-7.4, unpublished results). To determine the effect of HS on HSVEC morphology, the cells were fixed and stained with phalloidin, an actin stain.
Membrane integrity was determined by measuring ATP (Sigma, St Louis, MO) and lactate dehydrogenase activity (LDH, Cytotox-One, Promega, Madison, WI) release. In addition, transendothelial cell electric resistance (TEER) was measured with a voltohmmeter (World Precision Instruments, Sarasota, Fl), and STX2 electrodes (World Precision Instruments). HSVEC (passages 3-6) were plated (75,000 cells) in the polycarbonate membrane (0.4 µm) 12 mm transwell inserts in 12 well tissue culture plate (Costar, Corning, NY) coated with a solution of human fibronectin (50µg/ml in phosphate buffered saline, Sigma) The cells were cultured until the TEER was ≥20Ω.cm2) and treated with HS or HP for 2 hours and TEER was measured.
Intact rat aorta (RA) was collected from euthanized female, 250-300g, Sprague Dawley rats. The aorta was isolated via a midline incision and cut into 2-3mm rings and treated with HP, HS, or HS with apyrase (4U/ml ) or HS with A740003 (100µM) for 2 hrs at room temperature. Rings were suspended in a muscle bath containing a bicarbonate buffer, contracted with 110 mM potassium chloride, to determine smooth muscle functional viability, and then contracted with phenylephrine (PE, 0.5 µM) and relaxed with carbachol (CCH, 0.5 µM ), an acetylcholine analogue (Radnoti force transducer (model 159901A,Radnoti LLC, Monrovia, CA) interfaced with a PowerLab data acquisition system and Chart software (AD Instruments Inc., Colorado Springs, CO). Percent relaxation was calculated as a change in stress compared to the maximal PE-induced contraction which was set as 100%.
To determine the signaling pathways activated by HS injury, cells were homogenized, proteins separated by gel electrophoresis, transferred to a membrane, and stained with antibodies that recognized phosphorylated and non-phosphorylated p38 MAPK (Cell Signaling Technology, CA), and phosphorylated and non-phosphorylated MAPKAP kinase 2 (MK2, Cell Signaling Technology).
Results: HSVEC incubated in HS exhibited abnormal rounded morphology (Figure 1).
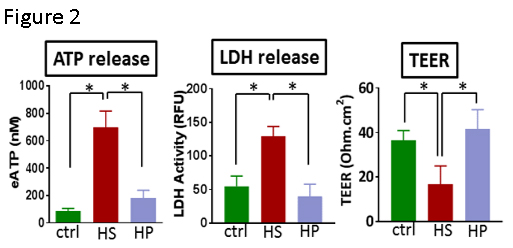
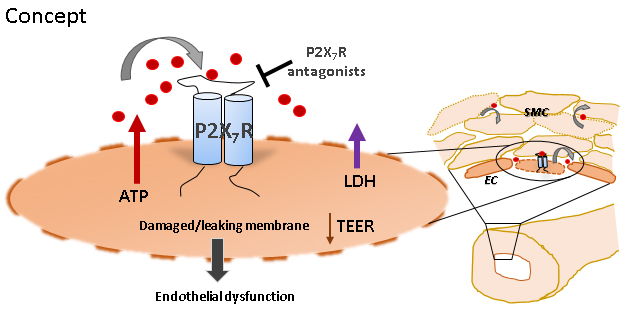
Previous studies have demonstrated that stretch injury of RA leads to release of ATP. The amount of ATP in the medium was significantly increased in HSVEC after storage in HS [696.9±119.3nM (HS) vs. *87.23±18.26nM (control untreated) and *182±55.74nM (HP); n=5, *p<0.05, Figure 2].
We postulated that storage in HS led to ATP release via membrane disruption. Treatment of HSVEC with HS led to increased LDH release [54.6±7.65 RLU (HS) vs. *111±19.15RLU (control untreated) and *39.14±8.41RLU (HP); n=3-5, *p<0.05, Figure 2].
Transendothelial cell electric resistance (TEER) is a measure of membrane integrity. HSVEC stored in HS had decreased TEER [16.83±3.33Ω.cm2 (HS) vs. *36.5±1.78Ω.cm2 (control untreated) and *41.5±3.6Ω.cm2 (HP); n=6, *p<0.05, Figure 2].
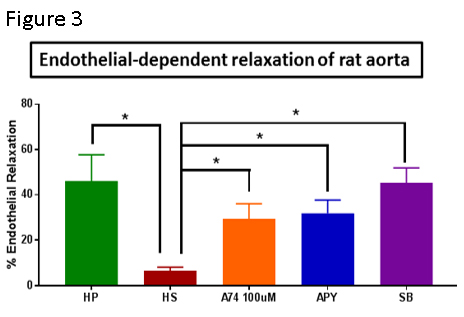
Since HS damages the endothelial membrane, we postulated that endothelial injury would lead to impaired endothelial function. Incubation of intact rat aorta (RA, a reproducible model of endothelial function in intact tissue) in HS impaired endothelial-dependent relaxation [6.41±1.64% (HS) vs .*46.1±11.7% (HP), n=8, p<0.05, Figure 3]. Unfortunately, HSV endothelial function in tissues obtained in the OR is impaired and could not be used as a model for these studies.
Since extracellular ATP leads to activation of a purinergic receptor (P2X7R), we determined if endothelial dysfunction could be restored by treatment with an enzyme that hydrolyzes ATP, apyrase (31.8±5.9%, n=8, p<0.05) and P2X7R inhibitor A740003 (29.4±6.7%, n=8, p<0.05), both of which restored endothelial function (Figure 3).
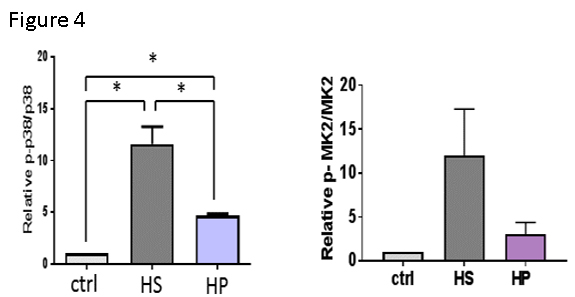
Finally, injury and P2X7R activation lead to activation of stress activated signaling pathways (p38 MAPK). P38 activates MAPKAP kinase 2 (MK2) which stabilizes cytokine mRNA and activates inflammatory pathways. To determine if injury and injury- induced activation of P2X7R in endothelial cells by HS leads to p38/MK2 activation, immunoblots were performed. Treatment of HSVEC with HS led to increases inP38 MAPK and MK2 phosphorylation (Figure 4).
To confirm that activation of p38 MAPK had a physiologic role in endothelial dysfunction after HS, RA were treated with HS in the presence of p38 MAPK inhibitor, SB203580. Co-treatment with SB203580 restored endothelial-dependent relaxation of RA (Figure 3).
Conclusions:
Incubation of HSVEC in non-buffered, acidic HS but not HP leads to morphologic changes and ATP release (Figures 1, 2). ATP is normally retained within the cytoplasm and a steep ATP concentration gradient exists between the cytoplasm (10-3 to10-2M) and the extracellular space (10-9 to 10-8 M). Thus, endothelial membrane damage would result in increased concentrations of ATP in the extracellular space.
To confirm that ATP release was due to membrane injury, LDH release and transendothelial cell electric resistance (TEER) were measured in HSVEC treated with HS. LDH concentrations in the supernatant increased and TEER decreased in HSVEC treated with HS, supporting the hypothesis that HS damages the endothelial membrane.
Treatment of intact RA with HS led to impaired endothelial function (Figure 3). To determine if endothelial dysfunction due to HS treatment was due to ATP release, RA was co-treated with HS and apyrase which hydrolyzes ATP. Apyrase treatment preserved endothelial function after HS injury (Figure 3).
P2X7 purinergic receptors (P2X7R), are activated by prolonged exposure to high concentrations of ATP (EC50 of 300-800µM). Treatment with the P2X7R inhibitor, A740003 also preserved endothelial function after HS injury (Figure 3). P2X7R are unusual in that ATP stimulation of P2X7R leads to ATP release from P2X7R. Thus, ATP activation of the P2X7R leading to further release of ATP represents a feedback process that may potentiate the response to injury.
Finally, injury and P2X7R activation are associated with activation of the p38 MAPK/MAPKAP kinase 2 (MK2) pathways. Treatment with NS led to increases in p38 and MK2 phosphorylation (Figure 4) and treatment of RA with the p38 MAPK inhibitor SB203580 prevented HS induced endothelial dysfunction (Figure 3) suggesting that these signaling pathways modulate the injury response to HS.
Activated MK2 modulates inflammation, cell migration and proliferation, and vascular remodeling. MK2-deficiency in mice prevents neointima formation, media hypertrophy, and lumen loss after wire injury of the common carotid artery. Our laboratory has developed a cell permeant peptide inhibitor of MK2 (MK2i) which inhibits intimal hyperplasia._ENREF_59 Activation of MK2 leads to phosphorylation of the ribonucleoprotein hnRNPA0 and tristetraprolin (TTP) which stabilize RNA for cytokine production (including IL-1β), resulting in activation of the pro-inflammatory response. IL-1β, is markedly induced in vessels after balloon injury and carotid ligation, and IL-1β signaling promotes vein graft thickening.
Taken together, these data suggest that injury-induced release of ATP, activation of the P2X7R/p38/MK2 signaling axis, and potentiation of the injury response via inflammation, represents a plausible and novel unifying hypothesis linking injury due to treatment with HS to vein graft failure. HP is an available option for graft storage that does not lead to endothelial injury. 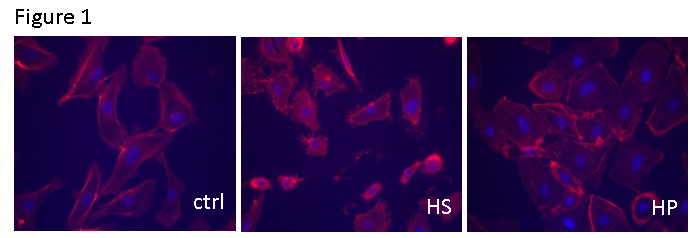
Back to 2018 Abstracts



